How does adaptive evolution operate at the genetic level in nature?
~ When a trait evolves, how many genes contribute?
~ Do the causative variants tend to reach 100% frequency?
~ Does natural selection usually act on new mutations or standing genetic variation?
~ Is adaptive evolution genetically predictable?
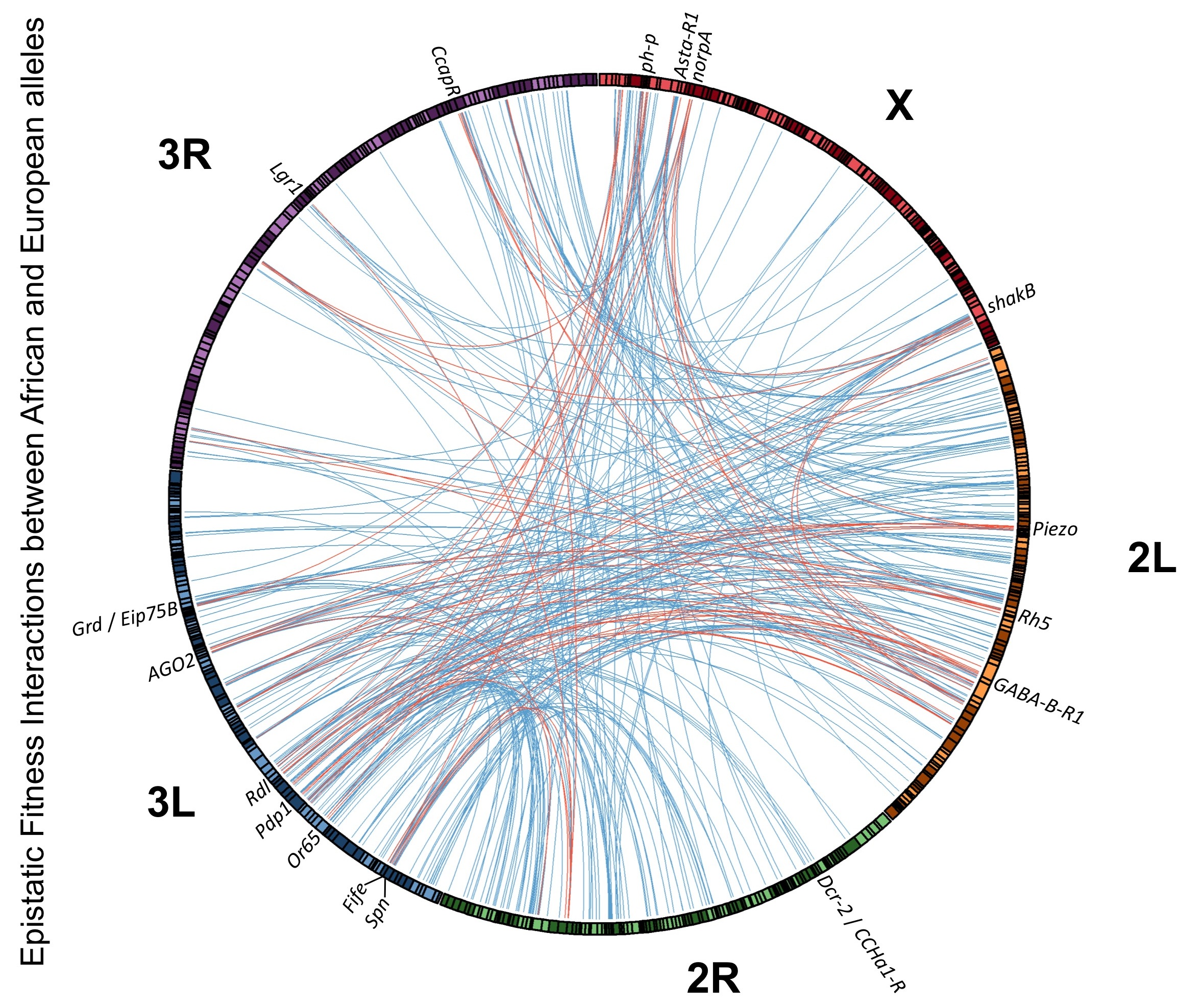
The Pool lab has a special emphasis on local adaptation (traits and underlying alleles that have a geographically limited advantage). Local adaptation gives us the clearest possible window into how adaptive evolution operates in nature: we can use crosses to map the causative genomic regions, we can analyze genetic variation to find differentially selected genes, and the unrivaled molecular tool kit of D. melanogaster can help to confirm and characterize the causative genes and variants. We follow just this pipeline to investigate the genetic underpinnings of a wide range of locally adaptive traits, including melanic pigmentation, large body and wing size, and elevated cold and ethanol tolerance. An emerging portrait from our studies is that selection may frequently act on standing genetic variants, including many of moderate to large effect, but the genetic architecture of these adaptive traits often remains variable within the evolved populations (Bastide et al. 2016; unpublished data). We are currently probing the potential role of epistasis among adaptive variants. We are also adding RNAseq data to add a transcriptomic dimension to our studies of local adaptation, which will bolster our understanding of how gene expression and alternative splicing contribute to adaptive change.
To complement our experimental efforts, we also maintain a strong focus on computational studies to extract new insights from genomic data sets, and developing new statistical methods to aid in these pursuits. Our recent and ongoing projects in this realm include (1) developing the Drosophila Genome Nexus (Lack et al. 2015; 2016) (2) new statistics for detecting local adaptation from genetic variation (Lange et al. 2016; Yassin et al. 2016), (2) a new method for analyzing bulk mapping data (Pool 2016), (3) a study investigating the impact of selective sweeps on the accuracy of demographic parameter estimates, (4) a novel hypothesis for the maintenance of polymorphic inversions, and (5) a temporal population genomic study that looks at the same natural population before and after 500 generations of evolution. Moving forward, we also aim to develop new methods to quantify local adaptation on a genome-wide scale, providing insights vital to both basic science and conservation.

How does one species begin to become two?
~ How quickly does reproductive isolation begin to develop between populations?
~ How important is natural selection in mediating admixture between populations?
~ Which genes and biological processes contribute to incompatibilities between populations?
Our interest in the earliest stages of speciation grows out of our studies of genomic diversity and local adaptation. Just as we discovered that a loss of developmental buffering can be one "cost of adaptation" (Lack et al. 2016), we likewise view the emergence of reproductive isolation between Drosophila populations as another potential side effect of adaptive divergence. Previous studies have shown that crosses between African and European D. melanogaster lead to lethality and sterility segregating among F2 offspring (indicating recessive incompatibilities) and that American populations of this species represent a mixture between these two gene pools. I previously showed that the North Carolina DGRP population shows patterns of admixture strongly influenced by natural selection, including a genomic abundance of "ancestry disequilibrium" between unlinked loci (Pool 2015). We are now working to identify the genes contributing to incompatibilities between African and European D. melanogaster, and we believe this will develop into a premier system for studying the genetic basis of reproductive isolation. Just as with our local adaptation research, we also want to infer the "big picture" models that are capable of generating the genomic patterns we observe.